 Corte histológico
de tejido cerebral afectado de encefalopatía espongiforme
Corte histológico
de tejido cerebral afectado de encefalopatía espongiforme
PRIONES
Los priones son algo nuevo y extraño para la sociedad actual, como siempre sucede en estos casos, el ciudadano medio recibe algún tipo de desconcertante información sobre un agente infeccioso cuando éste se ve inmerso en una noticia que conmociona a la sociedad por algún tiempo y luego se olvida tan rápidamente como llegó. Sin ir más lejos, los brotes recientes de Legionella en diversas regiones de España y, a escala más global, el mal que nos ocupa en esta sección, el conocido mal de las vacas locas, o como los científicos mejor lo definen, encefalopatía espongiforme bovina.
Pero, contrariamente a lo que pueda parecer en principio, la existencia de los priones se conoce desde los años 70. Y estos primeros descubrimientos van inevitablemente ligados a un nombre: Stanley Prusiner. La palabra prión deriva de "proteinaceous infectious particle", propuesta por Prusiner.
Los priones poseen un interés científico-sanitario, ya que no solamente son estudiados por su implicación en varios tipos de encefalopatías, sino que también nos aportan comprensión sobre los problemas biológicos desconocidos e importantes.
Características de las encepalopatías espongiformes transmisibles
enfermedades del encéfalo caracterizadas por la adquisición progresiva de un aspecto espongiforme (ahuecado), debido a la aparición de zonas de necrosis en diversas zonas del cerebro. Esta necrosis es producida por la acumulación de lo que se denomina placas de amiloide, formadas por una única proteína llamada la proteína del prión: PrP. La sintomatología de estas enfermedades es característica:
-periodo de incubación muy largo, de unos 30 años
-asintomáticas hasta que el daño es grande
-fase sintomática muy corta, al final de la enfermedad, que conduce a la muerte
-neurodegeneración progresiva: pérdida de funciones motoras, memoria etc
Existen encefalopatías espongiformes en hombres y animales como las ovejas y las vacas, de hecho, el mal tan famoso que afecta a las vacas se debe a un salto de especie desde las ovejas de la enfermedad llamada scrapie o prúrito lumbar, caracterizada por un picor insoportable en la columna vertebral de tipo nervioso que obliga a los animales a rascarse y contorsionarse de forma que parecen "locos".
Encefalopatías espongiformes en humanos
Creutzfeldt-Jacob (CJD): afecta al córtex cerebral y/o cerebelar y/o materia gris subcortical. Encefalopatía con la proteína del prion PrP. Inmunorreactividad, formación de placas o difusa. Tres formas: esporádica, iatrogénica (por causas médicas: riesgo reconocido en la neurocirugía) y familiar. Transmisión horizontal, componentes hereditarios en algunos casos y por ingesta (vacas locas).
Insomnio familiar fatal (FFI): degeneración talámica, espongiforme variable en cerebro. Ocurre en familias con la mutación PrP178 asp-asn.
Enfermedad de Gerstmann-Straussler-Scheinker (GSS): encefalo(mielo)patía con placas PrP multicéntricas. Ocurre en familias con ataxia dominante progresiva o con demencia.
Kuru: encefalopatía caracterizada por extensas placas de amiloide. Ocurre en la población de Nueva Guinea debido a los rituales de canivalismo
Corte histológico
de tejido cerebral afectado de encefalopatía espongiforme
La siguiente tabla resume las enfermedades del prión conocidas hasta ahora:
Enfermedad |
Huésped natural |
Prion |
Forma PrP anormal |
Forma PrP celular |
Scrapie |
Ovejas y cabras |
Scrapie |
ShePrPScç |
ShePrPSc" |
Prionencefalopatía transmisible del visón (TME) |
Visón |
prion TME |
MkPrPSc |
MkPrPTME |
Chronic wasting disease (CWD) |
Mulos, ciervos yAlces |
prion CWD |
MDePrPSc |
MDePrPCWD |
Encefalopatía espongiforme de los bovinos (BSE) |
Vacas |
prion BSE |
BovPrPSc |
BovPrPBSE |
Encefalopatía espongiforme de los felinos (FSE) |
Gatos |
prion FSE |
FePrPSc |
FePrPFSE |
Encefalopatía de los ungulados Exóticos (EUE) |
Nyala y el gran Kudu |
prion EUE |
NyaPrPSc |
NyaPrPEUE |
Kuru |
Humanos |
prion kuru |
HuPrPSc |
HuPrPKu |
Creutzfeldt-Jakob disease (CJD) |
Humanos |
prion CJD |
HuPrPSc |
HuPrPCJD |
Síndrome de Gerstmann-Straussler-Scheinker (GSS) |
Humanos |
prion GSS |
HuPrPSc |
HuPrPGSS |
Fatal familial insomnia (FFI) |
Humanos |
prion FFI |
HuPrPSc |
HuPrPFFI |
Propiedades de los priones
Físicas y químicas:
Filtrable con poros 25 nm o 100 nm. Es invisibles al microscopio óptico y electrónico. Resistente a:
·Formaldehido
· EDTA
· Proteasas ( Tripsina, pepsina ), aunque reducen la infectividad
· Nucleasas ( ribonucleasas A y III, desoxiribonucleasa I )
· Calor ( 360ºC)
· Radiación ultravioleta ( 2540 Å)
· Radiación ionizante
· Psoralenos e iones Zn
Biológicas
· Largo periodo de incubación ( meses, años, décadas).
· No producen respuesta inflamatoria
· No antigénicos.
· Patología crónica progresiva.
· Fatal en todos los casos.
· Carecen de cuerpos de inclusión.
· Presencia de ácido nucleico no demostrada.
· El único componente conocido es la proteína PrP.
· Pueden existir en múltiples formas moleculares
· Periodo de adaptación a nuevos hospedadores.
· Control genético de la susceptibilidad de algunas especies.
· Existencia de distintas cepas.
Los experimentos de inoculación en animales de laboratorio de la proteína PrP pura demostraron que ésta era capaz de transmitir la enfermedad sin necesidad de un ácido nucleico (DNA o RNA), cosa totalmente insólita en el mundo biológico, ya que hasta las formas más simples consideradas hasta el momento necesitaban de un ácido nucleico para poder ser infectivas (ejemplo: virus). Estas evidencias condujeron a Prusiner a la Teoría del prión: existen unos agentes infecciosos únicamente consistentes en proteína, sin ningún ácido nucleico asociado. Esto iba en contra de la biología molecular conocida y derrumbaba el dogma de un gen-una proteína (que actualmente podría considerarse más bien como un gen-una familia de polipéptidos isomorfos)
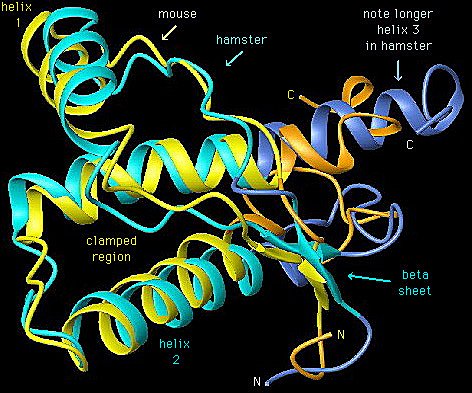
¿Pero cómo puede ser posible que una proteína tenga por sí misma naturaleza infecciosa? Los estudios rebelaron que una forma de la proteína PrP existe en la mayoría de los cerebros sanos de muchas especies animales: la PrPc sería entonces una forma de la proteína priónica no infectiva, ya que al inocularla en cerebros sanos no es capaz de desencadenar la enfermedad. Estas dos formas de una misma proteína contienen la misma secuencia de aminoácidos, pero su conformación espacial terciaria difiere; así, la forma infectiva, PrPsc, está formada por dos alfa-hélices y cuatro hojas-beta, mientras que la forma no infectiva (PrPc) contiene muchas alfa-hélices.
Los experimentos con animales knock-out para PrPc (diseñados genéticamente para que carezcan en su cerebro de la proteína PrPc ) demostraron que, a pesar de inocular la forma infecciosa, la enfermedad no se desarrollaba. Esto indicó que la proteína infecciosa necesitaba de la presencia de su forma sana para poder producir encefalopatía. Todo ello condujo a la conclusión de que el modo de actuación de la proteína priónica se basa en la inducción de cambios conformacionales de las formas sanas del cerebro hacia la forma infecciosa, algo así como el efecto que tiene la caída de la primera ficha en el efecto dominó. Es decir, que si por elemplo, se ingiere la proteína PrPsc desde carne infectada, ésta produce una reacción en cadena que se autoamplifica exponencialmente e irremediablemente en nuestro cerebro. Del mismo modo, las variantes hereditarias consisten en la mutación puntual de la forma sana hacia la forma infecciosa, siendo capaz una única mutación de extenderse por todo el encéfalo. Por ello las encefalopatías requieren un periodo de incubación tan largo, ya que se necesita tiempo para que la mayoría de las proteínas sanas sufran los cambios conformacionales necesarios y las acumulaciones necróticas para generar la enfermedad. También se ha observado una aparición espontánea de la conformación infecciosa de la proteína.
¿Qué características adquiere la proteína PrP al pasar a la forma infecciosa? El cambio conformacional confiere a la proteína gran resistencia a la degradación por proteasas, es decir, que se genera una alteración de su turnover y, al continuar habiendo una síntesis de proteína, ésta se acumula en el tejido cerebral, generando la necrosis. Además, el cambio la hace más insoluble, lo que facilita su precipitación y acumulación. Mientras que la forma normal se va degradando y regenerando equilibradamente, la forma infecciosa se acumula y cada vez genera el cambio de más formas sanas.
Pero, ¿cómo se produce este cambio conformacional? Las formas infecciosas PrPsc tienen una propiedad diferencial con respecto a las Prpc, la de actuar como una chaperona. Las chaperonas son proteínas que ayudan en el plegamiento de las demás proteínas. Las Prpsc actúan exclusivamente sobre las PrPc, induciendo su cambio conformacional secuencial.
Algo que mucha gente habrá pensado es ¿cómo puede ser que la ingesta de la proteína infecciosa induzca la enfermedad en el cerebro? Es bien sabido que el PH ácido del estómago es una barrera defensiva contra las infecciones, pero no es algo absolutamente infalible, de hecho, existe una bacteria capaz de sobrevivir a él (Helicobacter pylori) Es muy posible que la proteína del prión sea perfectamente capaz de "sobrevivir" al ambiente ácido del estómago e iniciar la ascensión hacia el cerebro a través de los nervios que llegan hasta la mucosa digestiva, ya que éstos también poseen la proteína PrPc; así, la forma infecciosa iría propagándose a través del nervio por una reacción en cascada de cambios conformacionales (el efecto dominó) hasta llegar al cerebro y una vez allí continuaría amplificándose.
¿Cómo llegaron a infectarse las vacas de una enfermedad de las ovejas? La hipótesis rebela que el origen del salto de especie podría deberse a un mal tratamiento de los piensos que ingieren estos animales, en principio elaborados con despojos de otros animales, incluidas las ovejas. Aunque la polémica incluya también el hecho de que se alimente a animales herbívoros con restos de carnes, vísceras etc, el problema no radica tanto en la composición de los piensos, sino en el cambio que se efectuó en el protocolo de elaboración de los mismos. De hecho, el inicio del brote coincide con un cambio en la temperatura de cocción de los despojos destinados a convertirse en harinas para el consumo animal. Quizás, al disminuir la temperatura a que se sometían estas harinas, se permitió la "supervivencia" en ellas de las formas infecciosas de la proteína priónica (probablemente porque la temperatura alcanzada era insuficiente para generar la desnaturalización de la proteína y su consecuente inactivación) De este modo, la proteína pasó de los despojos de las ovejas infectadas a las vacas que se alimentaban de ellos.
Presente y futuro de la encefalopatía espongiforme en humanos A mediados de la década pasada apareció una nueva variante de la enfermedad de Creutzfeldt-Jacob, caracterizada por un periodo de incubación más corto que la variante conocida. En estos nuevos casos, la enfermedad puede aparecer incluso a los treinta años de edad, lo cual es alarmante. Esta variante se ha asociado directamente a la ingesta de productos animales contaminados
El salto de especie, esta vez de la vaca al hombre, es debido a que la proteína priónica es capaz de actuar sobre sus formas sanas no homólogas (es decir, de otras especies) Debido a que muchos animales tienen PrP en su cerebro, es relativamente fácil que se de un salto de especie.
Actualmente no existe un test capaz de valorar qué individuos están infectados. El único análisis viable es post-mortem. Se desconoce el número de humanos que podrían estar incubando la enfermedad en el presente. Estos nuevos casos observados, ¿comprenden la totalidad de los individuos infectados recientemente o son una pequeña porción de la gran cantidad de enfermos que apareceran dentro de unos años (se ha calculado una aparición masiva hacia el 2015), generando una situación caótica sanitaria?
Nuevas incógnitas
-¿qué dosis de proteínas priónicas se necesita para desarrollar la enfermedad?en teoría, una única proteína es capaz de desencadenar el proceso patológico, debido a lo que hemos llamado en estas páginas "efecto dominó"
-la resistencia térmica poblacional de los priones es desconocida. Una cocción no da un 100% de seguridad.
-los animales sin sintomatología histopatológica, pero incubando la enfermedad, ¿pueden generar la patología en humanos al ser ingerida su carne?
-las normas actuales de separación de la médula ósea y otras partes de riesgo de la carne, ¿garantizan la no presencia de priones? ya que, la carne está inervada y los nervios pueden contener priones, sobretoto si la vaca se ha infectado por ingesta, lo cual quiere decir que los priones han viajado desde su estómago hasta el cerebro a través de los nervios, que recorren inevitablemente la carne considerada "pura" actualmente y, por tanto, apta para el consumo.
-¿la leche y los lácteos pueden contener priones?