GENES SUPRESORES DE TUMORES
Al igual que en el caso de la colaboración oncogénica, la pérdida de un solo gen supresor de tumores (incluso la pérdida de ambos alelos) no suele ser suficiente para causar cáncer. Su estudio resulta aún más complicado que el de los oncogenes, ya que siempre se haya más dificultad en el hecho de localizar la pérdida de un gen que en la localización de su presencia. En este sentido, el estudio del retinoblastoma, un cáncer humano poco común, ha resultado ser de gran utilidad. La pérdida del gen denominado Rb es el punto crítico para el desarrollo de este tipo concreto de cáncer. En pacientes con la forma hereditaria de la enfermedad, se presenta una delección o mutación de pérdida de la función del Rb en todas las células del cuerpo. Así, cuando se da la alteración del otro alelo en las células de la retina, aparece la enfermedad.
Existen varias formas posibles de perder ambas copias de un mismo gen:
De esta manera, cuando un individuo es heterozigótico para el gen, la alteración del alelo normal resulta mucho más frecuente que la alteración de ambos alelos cuando el individuo resulta ser homozigótico para la versión genética normal.
La siguiente tabla presenta una lista de los tipos de cáncer, cuyos genes implicados han sido identificados:
| TIPO DE CÁNCER | CONDICIÓN GENÉTICA | GEN |
| Cerebro | Síndrome de Li-Fraumeni Neurofibromatosis 1 Neurofibromatosis 2 Síndrome de von Hippel-Lindau Esclerosis tuberosa 2 |
TP53 NF1 NF2 VHL TSC2 |
| Mama | Cáncer hereditario de mama/ovario 1 Cáncer hereditario de mama/ovario 2 Síndrome de Li-Fraumeni Ataxia telangiectasia |
BRCA1 BRCA2 TP53 ATM |
| Colon | Poliposis familiar adenomatosa (FAP) Cáncer de colon hereditario sin poliposis (HNPCC) 1 Cáncer de colon hereditario sin poliposis (HNPCC) 2 Cáncer de colon hereditario sin poliposis (HNPCC) 3 Cáncer de colon hereditario sin poliposis (HNPCC) 4 |
APC hMSH2 hMLH1 hPMS1 hPMS2 |
| Endocrino (paratiroides,pituitaria, GI endocrino) |
Neoplasia múltiple endocrina 1 (MEN1) | MEN1 |
| Endocrino(feocromacitoma, tiroide medular) | Neoplasia múltiple endocrina 2 (MEN2) | RET |
| Endometrial | Cáncer de colon hereditario sin
poliposis(HNPCC) 1 Cáncer de colon hereditario sin poliposis (HNPCC) 2 Cáncer de colon hereditario sin poliposis (HNPCC) 3 Cáncer de colon hereditario sin poliposis (HNPCC) 4 |
hMSH2 hMLH1 hPMS1 hPMS2 |
| Ocular | Retinoblastoma hereditario | RB1 |
| Hematólogico(limfomas y leucemias) | Síndrome de Li-Fraumeni Ataxia telangiectasia |
TP53 ATM |
| Rińón | Tumor hereditario de Wilm Síndrome de von Hippel-Lindau Esclerosis tuberosa 2 |
WT1 VHL TSC2 |
| Pulmón | Historia familiar | Unknown |
| Ovario | Cáncer hereditario de mama/ovario 1 Cáncer hereditario de mama/ovario 2 |
BRCA1 BRCA2 |
| Próstata | Historia familiar | Unknown |
| Sarcoma | Retinoblastoma hereditario Síndrome de Li-Fraumeni Neurofibromatosis 1 |
RB1 TP53 NF1 |
| Piel | Melanoma hereditario 1 Melanoma hereditario 2 Síndrome de basal cell naevus (Gorlin) |
CDKN2 CDK4 PTCH |
| Estómago | Cáncer de colon hereditario sin
poliposis(HNPCC) 1 Cáncer de colon hereditario sin poliposis (HNPCC) 2 Cáncer de colon hereditario sin poliposis (HNPCC) 3 Cáncer de colon hereditario sin poliposis (HNPCC) 4 |
hMSH2 hMLH1 hPMS1 hPMS2 |
GEN RB
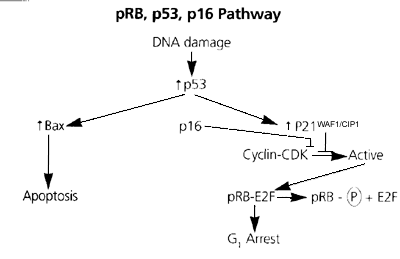
Este es el nombre del gen del retinoblastoma, aislado por primera vez en los 80, siendo a su vez el primer gen encontrado que causara cáncer humano. La pérdida de la función de RB causa el retinoblastoma hereditario, un tumor ocular desarrollado en los nińos cuyas células transportan una mutación desde la línea germinal. El desarrollo del tumor requiere dos mutaciones en las células precursoras embrionarias. RB juega un papel en la regulación del ciclo celular. Su producto en forma fosforilada puede unirse e inactivar el factor de transcripción E2F. También se une a una secuencia consenso de algunos promotores, inhibiendo la transcripción de los genes envueltos en el paso de la fase G1 a la fase S del ciclo celular. La proteína pRB es regulada por una un complejo ciclina quinasa-dependiente (CDK), quien fosforila pRB causando con ello su liberación de E2F, permitiendo a la célula continuar el ciclo celular. La pérdida de la pRB funcional se ha asociado a una gran variedad de procesos malignos en adición al retinoblastoma, ya sea durante las fases más tempranas o más tardías de su desarrollo.
La terapia génica ha sido utilizada para restaurar la copia funcional del gen RB tanto in vitro como in vivo. La terapia del RB también se beneficia de la capacidad de la pRB para incrementar la inmunogenicidad de las células tumorales y la supresión de la invasividad de las mismas.
GEN P16
El DNA del p16 fue aislado por vez primera en 1993 y se encontró que inhibía la actividad de la CDK (quinasa dependiente de ciclina), especialmente la del complejo D-CDK4. Esto colocaba al gen p16 en la misma vía que el gen RB. Mediante la unión a CDK4 y inhibiendo así la ciclina D, la p16 previene la fosforilación de la pRB, parando a la célula entonces en la fase G1. Mutaciones o deleciones de p16 han sido encontradas en una amplia variedad de tumores, sugiriendo su importante papel en el proceso de tumorgenicidad.
Se ha estudiado la eficacia del restablecimiento de la función de p16 mediante terapia géncia, resultando un efecto inhibitorio del crecimiento del tumor tanto in vitro como in vivo, así como una regresión del mismo en ciertos casos. Sin embargo, la eficacia del restablecimiento de la función del p16 podría limitarse a células cancerígenas con ciertos p16 y RB. Parece ser que la terapia del p16 resulta más citotóxica en células tumorales mutantes o nulas para p16, pero normales para RB, que para células que hayan perdido RB, pero que contengan p16 funcional.
Es preocupante el hecho de que el uso simultáneo de la terapia del p16 junto a quimioterapia parece conferir quimiorresistencia a las células cancerígenas. Otra complicación potencial es que p16 puede reducir la expresión del mRNA del gen RB y, por tanto, la de pRB. El mecanismo de este efecto no ha sido aclarado. Podría deberse en parte a una autorrepresión de RB, aunque ello no explicaría la totalidad del efecto observado. Se ha sugerido que la reducción en los niveles de pRB puede afectar a la terapia del p16, quizás requeriendo una co-introducción del gen RB junto al p16. Se ha encontrado que una sobreexpresión de p16 en el carcinoma hepatocelular se acompańa de un descenso total de los niveles de pRB. La apoptosis observada después del tratamiento con p16 iba ligada a una sobreexpresión de p53. Esto sugiere que el uso de p16 para bloquear la vía pRB normal, con el subsecuente descenso en la expresión de pRB, estimula la habilidad de la p53 sobreexpresada de inducir apoptosis. Así, los científicos esperan inducir la muerte celular tumoral vía p53 gracias a la terapia de p16. Aunque aún se necesita trabajar más en este campo para determinar el mecanismo concreto y judgar si los eventos descritos resultan ser universales o limitados a ciertos tipos tumorales o líneas celulares.
PAPEL DEL GEN SUPRESOR DE TUMORES P53 Y SU RELACIÓN CON LOS VIRUS
Es considerado como el mayor defensor de la célula . Alteraciones del gen p53 se ven envueltas, directa o indirectamente, en la mayoría de los cánceres humanos. El gen llamado p53 detiene el crecimiento de los tumores, pero su alteración se relaciona con el 60% de los cánceres.
En 1979, David Lane de la University of Dundee in Scotland y Arnold Levine de Princeton University descubrioeron independientemente la proteína p53, que codifica el gen con el mismo nombre. En 1982 los biólogos pudieron aislar el gen p53, pero no fue hasta 1989 cuando los científicos descubrieron su rol de gen supresor de tumores. Actualmente existen más de 5,200 publicaciones sobre p53. Algo que fascina a los biólogos es que el gen p53 permite predecir cómo tratar un cáncer.
Una célula sana suele contener un escaso número de proteínas p53, en degradación y renovación constante.Pero si la radiación ionizante, una carcinógeno químico, quimioterapia etc dańa el DNA, la célula entra en alerta y alguna seńal impide la degradación de p53, resultando en un incremento de la misma. P53 entonces se encarga de bloquear la maquinaria de división celular hasta que el dańo del DNA sea reparado y la célula pueda continuar su ciclo con normalidad. Su mecanismo de actuación es la unión a secuencias específicas del DNA, activando la expresión de ciertos genes cercanos, cuyas proteínas inhiben la división celular. Es habitual que cuando no se logre reparar la lesión del DNA, la misma p53 induzca a la célula a entrar en apoptosis.
La proteína p53 tiene esencialmente dos funciones:
Su estructura presenta lugares fácilmente alterables.
Al codificar para un factor de transcripción, la proteína p53 suprime la formación tumoral. Además, varios trabajos han postulado que podría estar comprometido en la reparación del DNA. Investigadores de la Universidad de Tokio, han reportado por primera vez que el gen p53 participa de manera directa en el proceso de reparación del DNA, al inducir la transcripción del gen p53R2.
Según los datos obtenidos, ésta última molécula codifica la subunidad R2 de la enzima ribonucleótidoreductasa (RNR, del inglés Ribonucleotide Reductase), que cataliza al producción de deoxirribonucleótidos trifosfato (dNTPs), necesarios para la replicación y reparación del DNA.
Así, ante una lesión del DNA, el gen p53R2 es activado por p53 y se une a la subunidad R2 de la enzima, permitiendo su paso al núcleo celular y el empleo de dNTPs para la producción de los correspondientes deoxirribonucleótidos, que son incorporados en los segmentos de DNA lesionado.
La inactivación de p53, reportada hasta en 80% de las neoplasias, impide la inducción de p53R2 y una respuesta adecuada ante dańo del ADN, lo que explicaría su papel en la génesis tumoral.
Las alteraciones del gen p53 suelen ocurrir por mutación puntual. Una de las 2.362 bases de su secuencia puede cambiarse por otra. Si esta mutación ocurre para una zona que codifique una región importante de la molécula, el efecto puede resultar en una anulación de su función como supresora de tumores. Las mutaciones del gen p53 pueden ser hereditarias, ya que abarcan a todo tipo de células, incluyendo a los gametos. En teoría, no debería causar problemas la heterozigosis para los genes p53 mutados, ya que el alelo con la versión no mutada podría ser capaz de producir suficiente cantidad de la proteína como para realizar su labor. Pero en realidad esto no ocurre, ya que los individuos heterozigotos para p53 son propensos a desarrollar varios tumores independientes en una amplia variedad de tejidos a edades tempranas. Esta predisposición familiar se denomina Síndrome de Li-Fraumeni.
Las dos versiones de la proteína p53, la salvaje y la del gen mutado, son capaces de acomplejarse en un tetrámero. Esto tendría un efecto devastador para la célula, ya que la proteína de la versión mutada estaría anulando a la versión normal y, por tanto, la célula se vería igualmente privada de su defensa contra la proliferación descontrolada. Las sustancias químicas, como el benzopireno del humo del tabaco, cambian las bases G por T y C por A y viceversa. Una sola mutación es capaz de provocar una célula potencialmente tumoral y una sola célula alterada es capaz de producir un tumor. Los cánceres causados por p53 mutadas son especialmente agresivos debido a que no solamente no evitan la división celular, sino que además tienden a estimularla. Es posible determinar las causas de un cáncer a partir del examen en una biopsia de la proteína p53. El diagnóstico basado en p53 es usado para la detección precoz del cáncer, así como para determinar la mejor terapia a seguir en cada caso concreto. En el caso de la radioterapia es esencial conocer el estado del gen p53, ya que se asume que ésta dańará las células ligeramente y activará al gen para que haga su función; el problema reside en el hecho de que un cáncer tenga alterada la función de p53, en tal caso la radioterapia no sólo no es útil, sino que es capaz de producir más dańo en las células del ya existente. Así, los tumores con un gen p53 normal, son más sensibles a tratamientos como la quimioterapia y la radioterapia que los que han mutado el gen.
Los oncólogos tienen puestas sus esperanzas en este gen y, según afirman, las posibilidades de tratamiento que ofrece sólo son limitadas por la propia imaginación. Un experimento se basaba en la utilización de virus como vectores portadores del gen p53 normal, dirigidos contra células cancerosas de nueve pacientes. En tres pacientes el experimento resultó en una regresión del crecimiento del tumor. Más sorprendente fue el hecho de que los vectores virales no tenían que infectar a todas las células para que se produjera el efecto. Este hecho se consideró como un efecto colateral de las células que sí fueron infectadas.
Este tema resulta ampliado en el apartado titulado como estrategias de la terapia génica con vectores adenovirales.