LA SINDROME DA ANTICORPI ANTIFOSFOLIPIDI
Angela Tincani & Marco Taglietti
Servizio di Allergologia e Immunologia Clinica,
Spedali Civili di Brescia
"Laborama" anno 4, n.° 4, 1999

STORIA
CRITERI DI CLASSIFICAZIONE
DIAGNOSI CLINICA e
DI LABORATORIO
LA STORIA
Storicamente la definizione di Sindrome da Antifosfolipidi (APS) risale a quando venne segnalata la positivitŕ del Lupus Anticoagulant (LAC) in pazienti con ripetute perdite fetali del periodo centrale della gravidanza, pazienti che avevano anche sofferto di episodi trombotici piů o meno ricorrenti ( 1,2).
Tuttavia la definizione del quadro nella sua interezza č largamente dovuta alle osservazioni cliniche che hanno accompagnato e seguito la introduzione del test degli anticorpi anticardiolipina nel 1983. A partire da quel momento, probabilmente grazie ai numerosi studi clinici certamente facilitati dalle caratteristiche del nuovo test, č risultata chiara la significativitŕ della associazione tra anticorpi antifosfolipidi (aPL), comunque rilevati, e un certo quadro clinico, caratterizzato da aborti ricorrenti, trombosi arteriose o venose e piastrinopenia (3). Questa associazione, appunto battezzata Sindrome da Antifosfolipidi, č stata in origine descritta con una certa frequenza nell'ambito del lupus sistemico, ma poco dopo č stata osservata anche in soggetti senza manifestazioni di altra malattia autoimmune. Pertanto č stata distinta la sindrome primaria nella quale i soli problemi sono quelli inclusi nella sindrome stessa, da quella secondaria, nella quale abortivitŕ, episodi trombotici o trombocitopenia sono soltanto una parte di un quadro clinico piů complesso(4, 5, 6).
NUOVI CRITERI DI CLASSIFICAZIONE
Recentemente, durante l'8° Simposio Internazionale sugli Anticorpi Antifosfolipidi, tenutosi a Sapporo in Giappone nell'ottobre 1998, č stato raggiunto un accordo internazionale sui criteri di classificazione. Lo scopo di questo sforzo era quello di fornire dei requisiti rigidi e inconfutabili per poter creare dei gruppi di pazienti assolutamente omogenei, adatti per la elaborazione di studi controllati. Si č pertanto privilegiata la individuazione delle caratteristiche essenziali della APS che emergono dalla osservazione degli studi clinici prospettici da un lato e dai modelli sperimentali dall'altro (tab. 1).
Tab. 1- Dichiarazione di Consenso Internazionale sui
Criteri classificativi della Sindrome da Antifosfolipidi
da Wilson W, Gharavi A, et al.; Arthritis Rheum. vol. 42, n.° 7 - luglio 1999
Richiesti: 1 o piů criteri clinici ed 1 o piů criteri laboratoristici nello stesso paziente.
| Criteri clinici |
1. Trombosi vascolari: uno o piů episodi di trombosi arteriose,
venose o dei piccoli vasi, in qualsiasi organo
o tessuto, confermate da tecniche di imaging, doppler
o dall' istopatologia. |
2. Patologia ostetrica:
a) Una o piů morti fetali oltre la 10^ settimana;
b) Uno o piů parti prima della 34^ settimana,
accompagnati da preeclampsia o severa insufficienza placentare;
c) Tre o piů aborti prima della 10^ settimana. |
| Criteri laboratoristici |
1. Anticorpi anticardiolipina (aCL) dipendenti di classe IgG e/o IgM
a titolo medio-alto, misurati con metodiche
ELISA standardizzata in due o piů occasioni
ad almeno 8 settimane di intervallo. |
2. Lupus Anticoagulant risultato positivo in due rilevazioni a 8 o
piů settimane di intervallo,
rilevato secondo il metodo raccomandato dal Sottocomitato
del Lupus Anticoagulant/phospholipid
Dependent Antibodies, consistente nei seguenti passaggi:
a) Prolungamento di un test di coagulazione dipendente
dai fosfolipidi (KCT, aPTT, DRVVT, ecc.);
b) Mancata correzione con mixing di plasma normale;
c) Correzione ottenuta con aggiunta di fosfolipidi;
d) Esclusione di altre coagulopatie. |
In effetti esistono studi prospettici che nel lupus e nell'aborto idiopatico mostrano una maggior frequenza di perdite fetali nella pazienti con anticorpi antifosfolipidi (7). Inoltre nei pazienti con trombosi venosa profonda, č stato dimostrato un rischio piů elevato di ricorrenza trombotica associato alla presenza di anticorpi antifosfolipidi (8).
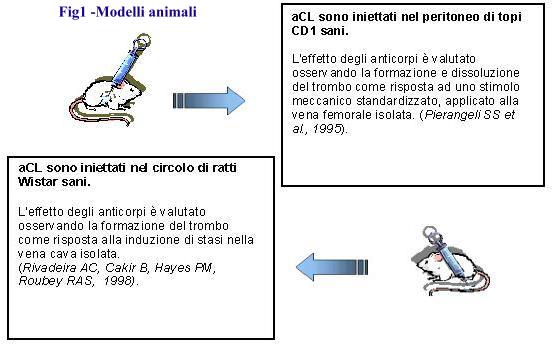
Anche i dati sperimentali sono in questo senso molto chiari dal momento che animali sani, infusi con anticorpi antifosfolipidi, sono soggetti ad una aumentata frequenza di aborti e, almeno in particolari situazioni, sviluppano trombosi o alterazioni della coagulazione (9) (vedi fig. 1).
Da quanto detto si dovrebbe dedurre che questi, come tutti i criteri classificativi, non sono destinati alla diagnosi del singolo paziente. In effetti sia nella parte clinica che in quella di laboratorio la diagnosi puň essere confortata da una serie di altre informazioni in qualche caso meno specifiche, ma pur sempre rilevanti, in altri casi semplicemente piů nuove quindi meno validate dalle osservazioni prospettiche cui precedentemente accennato.
LA DIAGNOSI
La diagnosi di APS deve necessariamente comprendere due approcci complementari e indispensabili: uno clinico ed uno laboratoristico.
DIAGNOSI CLINICA
Dalla analisi computerizzata delle caratteristiche cliniche di 667 pazienti con malattia lupica sono stati evidenziati i sintomi piů frequentemente associati ad aPL (10).
A conferma di quanto giŕ era stato evidenziato nelle casistiche piů ridotte riportate in letteratura tali sintomi sono:
- Perdite fetali ricorrenti
- Trombosi venose
- Trombosi arteriose
- Ulcere agli arti inferiori
- Livedo reticularis (fig. 2)
- Anemia emolitica
- Trombocitopenia
Le perdite fetali sono, nella nostra esperienza, il sintomo piů frequente che conduce allo studio e all'inquadramento delle pazienti. Come specificato nella tabella dei criteri, un "peso" diverso viene assegnato alle morti fetali (dopo le prime 10 settimane di gestazione), rispetto agli aborti precoci. Piů significative rispetto alla diagnosi sono considerate infatti le morti fetali, di cui una č sufficiente come criterio, rispetto agli aborti precoci dei quali sono richiesti almeno tre. Anche la preeclampsia ed il ritardo di crescita, considerati criterio classificativo, rientrano nei problemi del II e III trimestre, per cui la Sindrome da Antifosfolipidi nel suo insieme, pare essere piů frequentemente un problema dello sviluppo fetale e, meno frequentemente, un problema di impianto dell'embrione.
La patologia ostetrica associata ad APL puň avere, almeno in parte, origine da fatti trombotici. In effetti, fatti trombotici a livello dei vasi placentari sono stati descritti frequentemente. Verosimilmente tuttavia, altri meccanismi patogenetici, come una azione piů diretta degli APL sul trofoblasto, sembra possano essere implicati nel danneggiamento del feto, talvolta sproporzionato rispetto ai fenomeni trombotici rilevati a livello placentare.
Nella sindrome si riscontrano trombosi a carico del circolo venoso ed arterioso. I vasi piů frequentemente riportati come sede di trombosi venose sono le vene profonde e superficiali degli arti inferiori.
In queste situazioni sono stati osservati episodi di embolia polmonare che se ripetuti possono essere causa di ipertensione polmonare. E' comunque utile ricordare che le trombosi venose nell'ambito della sindrome sono potenzialmente ubiquitarie e in effetti sono state riportate in distretti diversi condizionando pertanto quadri clinici correlati all'organo danneggiato e quindi assolutamente disomogenei.
La sede piů frequentemente descritta come colpita da trombosi arteriose č quella cerebrale. Il quadro riportato č usualmente quello dell'infarto cerebrale con il danno relativo all'area encefalica danneggiata. Tuttavia taluni infarti cerebrali possono decorrere in modo asintomatico o, all'opposto, possono essere cosě estesi da presentarsi addirittura come demenza o epilessia. Come per le trombosi venose anche per quelle arteriose vale il concetto della possibile ubiquitarietŕ di sede del fenomeno e conseguentemente di una presentazione clinica correlata al sito anatomico compromesso. La giovane etŕ del soggetto affetto e la assenza di fattori di rischio per l'arteriosclerosi possono far sospettare la diagnosi di APS.
Benché la trombosi dei vasi di grosso calibro sia di piů frequente osservazione, anche i vasi di calibro medio piccolo possono essere implicati nella sindrome. Sono descritti infatti casi in cui la trombosi delle arterie interlobulari e delle arteriole e capillari glomerulari condizionano una alterazione della funzione renale, caratterizzata da proteinuria, ipertensione e talvolta insufficienza renale. Inoltre, nel derma, fatti trombotici a livello arteriolare possono causare lesioni ulcerative, macule eritematose, porpora o fatti necrotici. In questi casi la diagnosi č ovviamente istologica ed č caratterizzata da emorragie e depositi di emosiderina attorno ai vasi danneggiati, oltre che ovviamente da mancanza di infiltrato flogistico. Pertanto anche le ulcere degli arti inferiori a lenta risoluzione, che si associano significativamente alla sindrome, sono su base trombotica e rientrano pertanto anche nei criteri classicativi.
Altre manifestazioni associate in modo significativo alla sindrome come la livedo reticularis (dovuta a un rallentamento del circolo nei vasi del derma), la anemia emolitica e la trombocitopenia non sono su base trombotica e, pertanto, pur essendo suggestive per una diagnosi non sono ritenute oggi criterio classificativo.
Naturalmente la rilevazione di questi sintomi fa sospettare la APS e pertanto avvia le procedure laboratoristiche richieste, solo quando le cause piů comuni siano state escluse.
Oltre a queste caratteristiche, basandosi su osservazioni abbastanza condivise anche se non controllate, altri sintomi sono stati descritti come associati ad APS. Tra questi pricipalmente la malattia valvolare cardiaca e un certo numero di manifestazioni neurologiche non sempre o non chiaramente riconducibili a trombosi (ischemia cerebrale transitoria, mielite trasversa, corea, emicrania).
Pertanto data la riconosciuta frequenza di queste caratteristiche, anche se non rivestono carattere diagnostico, se ne raccomanda la rilevazione che puň confermare un sospetto diagnostico o stimolare alla effettuazione di accertamenti strumentali che in alcuni casi potrebbero portare alla dimostrazione di fatti trombotici.
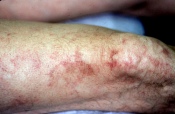 Figura 2 - Livedo reticularis
Figura 2 - Livedo reticularis
DIAGNOSI DI LABORATORIO
A differenza di molte altre patologie autoimmuni, la diagnosi laboratoristica di APS puň comprendere anche la positivitŕ di un solo test. Per questa ragione viene raccomandato estremo rigore nella valutazione dei test diagnostici e nel controllo della loro persistente positivitŕ.
TESTS CLASSICI
E' evidente che nel rispetto del consenso internazionale sui criteri classificativi della APS i tests diagnostici sono il Lupus Anticoagulant, test di coagulazione fosfolipidi-dipendente descritto giŕ negli anni '50 (vedi tabella precedente) e il test per gli anticorpi anticardiolipina, purchč siano rigorosamente rispettate le modalitŕ sopra riportate.
a) LUPUS ANTICOAGULANT
Seguendo le linee guida elaborate dal Comitato Scientifico e di Standardizzazione della Societŕ Internazionale per la Emostasi e la Trombosi (11), la ricerca del LAC, viene condotta secondo una procedura che prevede tre tappe. In primo luogo č necessaria la dimostrazione di un significativo prolungamento di (o dei) test di coagulazione fosfolipidi-dipendenti: i tests piů diffusi comprendono la determinazione del Tempo di Tromboplastina Parziale Attivata (aPTT), invero non particolarmente sensibile, del Tempo di Coagulazione al Caolino (KCT) e il test al Veleno di Vipera Russel diluito (dRVVT). Qualora i tempi di coagulazione risultino superiori a quelli di normalitŕ, diviene necessario escludere la possibilitŕ di un eventuale deficit di fattori della coagulazione nel campione in esame, ripetendo i test su un miscela del campione in esame e di plasma normale: la mancata correzione dei tempi di coagulazione depone per la presenza di anticorpi ad attivitŕ anticoagulante. L'ultima tappa (test di conferma) consiste nella dimostrazione che la aggiunta di fosfolipidi (lisato piastrinico o fosfolipidi a conformazione esagonale) č in grado di correggere il tempo di coagulazione prima prolungato.
b) ANTICORPI ANTICARDIOLIPINA
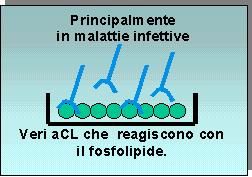
Il test degli anticorpi anticardiolipina č stato descritto nel 1983 come metodo radioimmunologico ma č stata rapidamente trasformato in metodo immunoenzimatico (ELISA) e come tale č oggi eseguito nel mondo. Durante il processo di standardizzazione della metodica avvenuto negli anni 80 era emerso chiaramente un accordo generale sull'utilizzo di siero bovino fetale o adulto come componente della soluzione tampone utilizzata per il bloccaggio e per la diluizione dei campioni in esame. L'uso del siero bovino, rispetto alla gelatina e/o all' albumina bovina precedentemente impiegati, consentiva infatti un notevole miglioramento delle prestazioni della metodica in termini di binding piů elevato, riproducibilitŕ, etc. Tale scelta privilegiava evidentemente solo taluni aspetti della metodica da standardizzare, trascurando sostanzialmente la presenza nel siero di proteine in grado di legarsi ai fosfolipidi a carica negativa, e quindi alla stessa cardiolipina copulata alle piastre. Questo ha comportato che con la metodica cosě standardizzata (aCL ELISA classico, fig. 3) fossero rilevabili come antigene non solo la cardiolipina, ma anche proteine sieriche ad essa adese, con conseguente perdita della specificitŕ. In effetti nel 1990, tre gruppi di ricerca constatarono indipendentemente che il legame di anticorpi aCL altamente purificati richiedeva una componente del siero o del plasma. Il componente in questione, denominato cofattore della cardiolipina, fu identificato nella beta 2 glicoproteina I.
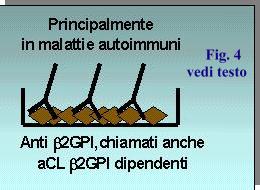
Le conoscenze acquisite negli ultimi 10 anni hanno evidenziato come i cosě detti anticorpi anti fosfolipidi siano diretti in larga parte non verso il fosfolipide di per sé ma piuttosto verso le proteine che legano i fosfolipidi (es.: beta2GPI, protrombina etc.). In particolare la beta2GPI č stata riconosciuta come parte principale del complesso antigenico riconosciuto dagli anticorpi anti cardiolipina. Per questo motivo sono stati sviluppati dei metodi ELISA per il rilevamento di anticorpi anti beta2GPI che rivestono oggi un significato diagnostico apparentemente piů specifico e piů sensibile del test classico degli anticorpi anti cardiolipina (12) (Fig. 4). Pertanto anche se questo metodo non č entrato in quelli approvati dal consenso internazionale, probabilmente anche per la attuale carenza di studi di standardizzazione del metodo, un paziente con sospetto clinico di APS e anticorpi anti-beta2GPI, anche in assenza di positivitŕ del LAC o degli aCL ha una forte probabilitŕ di avere la sindrome. Il giudizio finale resta al medico che deve tenere in considerazione in questo anche la affidabilitŕ del laboratorio o dei laboratori che forniscono i dati.
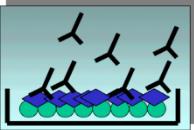 Fig. 3 - aCL ELISA classico
Fig. 3 - aCL ELISA classico

TESTS COMPLEMENTARI
Essendo la APS una malattia autoimmune sistemica la positivitŕ di altri autoanticopi č tutt'altro che rara e puň servire a confermare o completare la diagnosi.
Tests descritti nella APS con varia frequenza, in grado di confermare la diagnosi sono rappresentati da: Test di Coombs diretto, anti-mitocondrio di tipo M5 (AMA-M5), anticorpi anti lamine.
Al fine di poter completare la diagnosi, sono stati poi individuati vari parametri, quali:
- I tests per la valutazione degli anticorpi antinucleo, che puntualizzano la eventuale relazione tra la APS con una malattia Lupus Like o addirittura la sua secondarietŕ a Lupus Eritematoso Sistemico (ANF con titolo, anticorpi anti ENA, anticorpi anti DNA, nativo e denaturato);
- La valutazione del sistema complementare, sia per meglio definire la relazione della APS col Lupus Eritematoso, ma anche per rilevare eventuali deficit di fattori complementari descritti nella APS (CH50, C3, C4);
- Indagini routinarie, in particolare:
Emocromo completo con formula e piastrine (per la valutazione della eventuale piastrinopenia e anemia);
Funzionalitŕ renale;
Funzionalitŕ epatica;
Test di flogosi che dovrebbero essere negativi nella APS (salvo eccezioni: una potrebbe essere la VES elevata in caso di anemia emolitica Coombs' +).
Come specificato nei criteri diagnostici, la diagnosi di APS prevede l'esclusione di altre coagulopatie, pertanto i pazienti dovranno essere indagati per le piů comuni cause di trombofilia (da concordarsi con il laboratorio di coagulazione).
Bibliografia
1. Alagille D, Crossnier J, Sullier PJ; 1956.
2. Nilson, IM, Astedt, B, Hender, U; 1975.
3. Harris EN, et al.; Br. J. Rheumatol. 1987; 26:324-7
4. Asherson, RA; J Rheumatol. 1988 Apr;15(4):539-43.
5. Font,J, Cervera, R; Med Clin (Barc). 1988 Mar 26;90(12):490-3.
6. Alarcón-Segovia, D, Sanchez-Guerrero; J Rheumatol. 1989 Apr;16(4):482-8
7. Lockshin MD,et al. N Engl J Med 1985; 313: 152-6
8. Ginsberg et al, Blood 1995; 86: 3685-91.
9. Tincani A et al., Expansion Scientifique Publications, 1998
10. Alarcón-Segovia et al.; Sem. Arthritis Rheum. 1992; 21:275-285
11. Brandt JT et al.; Thromb Haemost 1995; 74:1185-90.
12. Tincani et al.; Clin Exp Rheumatol. 1998; 396-402.
 Pagina principale
Pagina principale
|

|

