Sameer Al-Arrayed, MRCP**
Ratnakar KS, MBBS,MD***
While extensive studies are available on the renal manifestations in sickle cell disease, there is no documented evidence in favour of renal changes in the commonly encountered haemoglobinopathy namely, ? thalassaemia. A light, immuno-flourescence and electron microscopic observations on a renal biopsy of a young male patient who is a known case of Hemoglobin H disease with proteinuria is documented. This is probably the first case reported, primarily for renal complications with demonstrable podocyte abnormalities. The pathogenesis of renal manifestations remains obscure. The renal alterations are less likely to be due to minimal change disease in view of immunoglobulin deposits in the mesangial area. However involvement of any other immune process or continued sub- clinical haemolysis producing proteinuria remain to be seen on long-term studies.
Bahrain Med Bull 2001;23(2):95-97.
Among the haemoglobinopathies, the renal complications
of sickle cell disease are well characterized1. The principal manifestations
include haematuria, sometimes profuse, hyposthenuria, nephrotic syndrome
and renal failure1,2,9. Pathogenetically, nephrotic syndrome remains speculative.
Alpha thalassaemia, although a common haemoglobinopathy, has not been reported
to be associated with proteinuria or other renal manifestations.
In view of the absence of reported literature on renal involvement in Haemoglobin-H
disease, a recently encountered case of proteinuria in
a patient with Haemoglobin-H disease in Salmaniya
Medical complex, Bahrain, is being reported and relevant literature is
reviewed.
THE CASE
A 38-year-old male from the eastern Saudi Arabian province was admitted for evaluation of dysuria, proteinuria of 18 months duration. There was no past history of loin pain, skin rash, arthritis, and oedema or drug allergy. There was also no past illness record of skin or throat infection or hemolysis or vaso-occlusion related renal crisis.He was not diabetic or hypertensive.He was taking tab.Losartan, prescribed elsewhere,for reducing proteinuria. On examination, patient was afebrile, pulse rate 86/mt. and regular, blood pressure 151/81 mm of mercury. There was mild splenomegaly.Rest of the clinical examination was unremarkable. Urinalysis revealed
proteinuria++, Hb+++ RBC-0-1/high power field,WBC-1-2/high power field and granular cast.Urine was sterile on culture. 24hr. proteinuria was between 0.7and1.2gm.Haemoglobin was11.3gm/dl, hematocrit 0.4,red blood cell count 5.7x1012/l mean cell volume 67fl, mean cell haemoglobin 29pg,mean cell haemoglobin concentration 29gm/dl,white blood cell count 5.2x109/l (P52,L32,M8,E3%) , platelet count 191x109/l,reticulocyte count was 6.0%,.The peripheral smear of blood showed microcytic hypochromic red cells and no schistocytes were seen. Hemoglobin H inclusions were demonstrable by staining with brilliant cresyl blue and incubating at 370C for 2hrs. G6PD activity was reduced. Sickling test was negative. Hb- Electrophoresis revealed Hb A/H (a-thalassemia), HbA2 was1.5%, Hb F was 0.2%, thus establishing the diagnosis of HbH disease. Blood glucose was 5.7mmol/l, urea 4.3mmol/l, creatinine 84 umol/l, sodium 142 mmol/l, potassium 4.3mmol/l, chloride 101 mmol/l, bicarbonate 29mmol/l, calcium 2.34 mmol/l, phosphorus 1.1mmol/l, serum total protein 83 gm/l, albumin 42 gm/l, bilirubin 23 umol/l (direct. 5, indirect 18), alkaline phoshatase 53u/l, alanine aminotransferase 64 u/l, gammaglutamyl transpeptidase 50 u/l, serum iron, vitamin B12 and folate were normal, C-reactive protein less than 10mg/l, erythrocyte sedimentation rate 2mm in the first hour. Anti-nuclear antibody, anti-double stranded DNA antibody, Hepatitis B surface antigen, Anti Hepatitis C Virus antibody and anti-HIVantibody were all negative. Immunoglobulin and complement profile were essentially within normal range. Prothrombin time was 19 (control l5), and activated partial thromboplastin time was 33 (control 28).


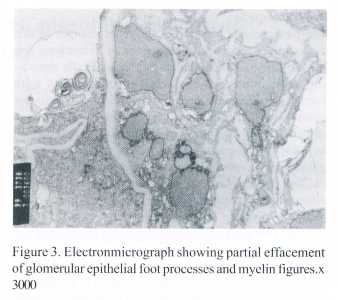
A percutaneous renal biopsy was carried out to know the cause of proteinuria. The biopsy showed 12 glomeruli with moderate enlargement and widened glomerular capillary lumina. There was focal distension of peripheral capillary loops [Fig1]. The red blood cells were seen discretely as well as in groups at the base/stalk of the glomerular tuft. The epithelial and mesangial cells were within normal limits. The capillary endothelial cells appeared edematous. Basement membrane was unremarkable. The proximal tubular epithelial cells in scattered areas showed intracellular haemosiderin pigment [Fig 2]. Blood vessels and interstitium were essentially normal. Immunoflourescence studies revealed diffuse fine granular staining for IgM in the measangium, weak or minimal fine granular staining for IgG, IgA and C3 in the mesangium of few glomeruli. Electron microscopy revealed normal thickness of basement membrane; epithelial cells were seen to have partial effacement of foot processes , electron dense granules (hemosiderin) and myelin figures of questionable importance [Fig3]. Endothelial cells were normal but capillary lumina were congested with red cells. Mesangium showed few small electron-dense deposits.
DISCUSSION
Alpha thalassaemia may be the most common single gene disorder in the world3. Its incidence has been reported as 24% in Bahrain10. Hemoglobin H disease, (with ? tetramer hemoglobin) is the most important clinical form of ? thalassaemia3, and is a disorder with varied clinical and haematological severity featuring chronic haemolytic anaemia, variable splenomegaly and bone changes. It is most frequently found in south-east Asia and the mediterannean region.. Molecular genetics and chain analysis allow us to precisely identify individuals and the families affected by the disorder. There are many oriental forms to confuse the issue both clinically and genetically3-5.
Identification of Hemoglobin H, the rapidly migrating hemoglobin on cellulose acetate gel electrophoresis at alkaline pH and the demonstration of haemoglobin H (HbH) inclusions in red blood cells after brilliant cresyl blue staining are diagnostic parameters of this disorder. The rapidity with which HbH forms hemichromes distinguishes the disorder from typical HbA and conversely HbBarts hemichromes are distinctly slower than HbH6. It has been documented that the incidence of a - thalassaemia is strikingly large in Saudi Arabia and the levels of HbBarts are highest in the world7. Weatherall et al considered that although haemoglobin H disesase(of aa T Saudi non-deletional form) has been reported in the oasis population of eastern Saudi Arabia, it is relatively rare4. The renal manifestation with proteinuria and the morphology of kidney showing immunoglobulin deposit and glomerular epithelial foot process fusion have not been reported hitherto in HbH disease to the best of knowledge of the authors. Glomerular enlargement seen in sickle cell nephropathy was also noted in this biopsy. However, unlike sickle cell nephropathy, morphological forms like focal segmental glomerulosclerosis or membrano-proliferative glomerulopathy to account for proteinuria were not seen in our case. In sickle cell disease, few cases of immune-complex nephropathy have been reported12, although it is uncertain whether this is part of sickle cell nephropathy or sickle cell disease modified nephropathy.
The pathogenesis of some of the renal changes in this case appears to be the consequence of haemolysis following clogging of capillaries by rigid red cells containing inclusions8. The tubular epithelial ells with haemosiderin indicate chronic intravasular haemolysis due possibly to HbH disease and G6PD deficiency. Although evidence of chronic intravascular haemolysis may be seen in kidney in several other haemolytic disorders, similar glomerular morphology has not been reported. The partial foot process fusion on electronmicroscopy indicates either primary disturbance or secondary to proteinuria itself. It is possible that this patient had a renal limited immune process with immune deposit in the mesangium causing proteinuria. However the contribution of hyperfiltration in the enlarged glomeruli to proteinuria cannot be ruled out. Interestingly, Guasch et al noted increased permeability in the glomerular basement membrane in sickle cell nephropathy11. Similar mechanism for proteinuria could be operating in the present case .The cause of immune deposits in the mesangium and the relationship of these deposits to proteinuria whether causal or incidental remains speculative and hence the natural course of the disease is obscure. Prospective studies are required to assess the incidence and the types of renal involvement in Haemoglobin H disease.
REFERENCES
1. Bucklewvm Jr, Someren A. Renal manifestations
of sickle cell disease. Arch
Intern Med 1974;133:660-9.
2. Bucklewvm Jr. The kidney in sickle cell
disease. The kidney: New York National
kidney foundation,1978;2:11-14.
3. Weatherall DJ, Clegg JB. The alpha thalassaemias.
In:The Thalassaemia
syndromes. London:
Blackwell scientific publications, 981:604-6.
4. Weatherall DJ. The Thalassemias.
In: William J, Bentler E,(eds). Hematology.
Chap 50,
3rd edn. London: McGraw-Hill Book Co,1983:493-521.
5. Jasim N, Al-Arrayed S, Gerard N, et
al. A mismatched-primer polymerase chain
reaction fragment
length polymorphism strategy for rapid screening of the
polyadenylation
signal mutation alpha (T-Saudi) (AATAAA?AATAAG) in the
alpha 2-globulin
gene. Haemoglobin 1999;23:213-20.
6. Rachmkewitz EA. Formation of hemichromes
from oxidized haemoglobin-
subunits. Ann
NY Acad Sci 1969;165:171-5.
7. Pembrey ME, Weatherall DJ, Clegg JB,
et al. Haemoglobin Barts in
SaudiArabia. Brit
J Haematol 1975;221-8.
8. Nathan DG, Gunn RB. Thalassaemias: The
consequences of unbalanced
haemoglobin synthesis.
Amer J Med 1966;41:815-30.
9. Saborio P. Sickle cell nephropathy.
J Am Soc Nephrol1999;10:192-9.
10. Mohammed AM, Al Hilli F, Nadkarni KW,
et al. Haemoglobinopathies and
G6PD deficiency
in hospital birth in Bahrain. Ann Saudi Med 1992;12:536-9.
11. Guasch A, Cua M, You W, et al. Sickle cell
anaemia causes a distinct pattern of
glomerular
dysfunction. Kidney Int 1997;51:826-33.
12. Pardo V, Strauss J, Kramer H, et al.
Nephropathy associated with sickle cell
anaemia. An
autologous immune-complex nephritis. Am J Med 1975;59:650-9.
--------------------------------------------------------------------------------
* SeniorResident, Nephrology
** Consultant Nephrologist
Department of
Medicine
*** Consultant Pathologist
Department of
Pathology
Salmaniya Medical
Complex
State of Bahrain
