Theory: As per the LCAO-MO theory, the electronic wavefunction Yel of a molecule is assumed to be an antisymmetrised product of the occupied spin-orbitals (SOs) formed out of the occupied molecular orbitals (MOs). Here each of the MO is assumed to be a linear combination (LC) of atomic orbitals (AOs) corresponding to the constituent atoms. However, in the H2+ molecular ion there is only one electron, and so here the electronic wavefunction Yel is identical, in its spatial part, with the lone occupied MO. This means that here the purely electronic energy Eel is same as the MO energy e of that MO.
As per the simplest MO theory of the H2+ ion (also of the H2 molecule), the two lowest-energy MOs are assumed to be linear combinations of the two 1s AOs i.e., the 1sa & 1sb AOs centred, respectively, on the two H nuclei a & b: thus the MO f equals (ca 1sa+cb 1sb). However, it would be grossly unrealistic to think of these contributing AOs as simply those of the contributing H-atoms: each contributing AO must have a (non-unity) orbital exponent (i.e., effective nuclear charge) k {i.e., 1sa = k3/2 p–1/2 exp(–kra) and 1sb = k3/2 p–1/2 exp(–krb) in atomic units} so that each generated MO f shows proper limiting behaviour as the internuclear distance R tends to zero (transforming the H2+ species to He+ atomic ion having nuclear charge 2), or tends to infinity (separating one H-atom from an H-nuclei). Thus the exponent k must be a function of the distance R: k must equal 2 as R equals 0, must equal 1 as R equals infinity, and be somewhere in between 1 and 2 for intermediate values of R, such as the distance corresponding to the given molecular ion.
Solving the secular equation that arises from the
linear variation theory treatment for this LCAO-MO problem,
we get the approximated energy e1 of the 1st (i.e., lowest-energy) MO
as e1 = (Haa+Hab)/(1+Sab). This MO
itself is f = ca(1sa+1sb).
Here Haa,
Hab and Sab are defined as Haa = ∫1saĤel1sadv,
Hab = ∫1saĤel1sbdv
and Sab = ∫1sa1sbdv, the dv = r2sinq dq.df.dr being
the volume element and Ĥel = –0.5![]() +1/ra+1/rb
(in atomic units) being the purely electronic Hamiltonian. As here the MO energy
e1
equals the purely electronic energy Eel,
so we get the electronic energy U (inclusive of internuclear repulsion VNN)
is U (= Eel+VNN) = (Haa+Hab)/(1+Sab) + 1/R
(here VNN
= 1/R -- in atomic units). From the form of the nuclear Schrodinger equation, we
known that this
energy U is also called the internuclear
potential energy deciding the nuclear motions. Now, performing the three integrations defined above, it can be
shown that the aforesaid integrals are expressible by the following simple
relations, finally leading to values of U as a function of only R and k {noting that U =
(Haa+Hab)/(1+Sab) + 1/R, as mentioned
above}:
+1/ra+1/rb
(in atomic units) being the purely electronic Hamiltonian. As here the MO energy
e1
equals the purely electronic energy Eel,
so we get the electronic energy U (inclusive of internuclear repulsion VNN)
is U (= Eel+VNN) = (Haa+Hab)/(1+Sab) + 1/R
(here VNN
= 1/R -- in atomic units). From the form of the nuclear Schrodinger equation, we
known that this
energy U is also called the internuclear
potential energy deciding the nuclear motions. Now, performing the three integrations defined above, it can be
shown that the aforesaid integrals are expressible by the following simple
relations, finally leading to values of U as a function of only R and k {noting that U =
(Haa+Hab)/(1+Sab) + 1/R, as mentioned
above}:
Overlap integral Sab = (1 + k*R + k*k*R*R/3) *
exp((-1)*k*R)
Coulomb integral Haa = 0.5*k*k - k - 1/R + (k+1/R) *
exp((-2)*k*R)
Resonance Integral Hab = (-0.5)*k*k*Sab - k*(2-k)*(1+k*R)*exp((-1)*k*R)
Now the value of the optimum orbital exponent k for any given distance R is obtained by again applying variation theory, this time numerically, as the value that would result in the minimum possible value of U for the given distance R. This has been done here using a small computer-program (enclosed herewith) written in Visual FoxPro 6 programming environment, in which the exponent k is varied within 1 and 2, first in steps of 0.01 and then in steps of 0.0001, and the value of k that has resulted in the minimum value of U is identified. The program then repeats this procedure for another value of the distance R. The program output displaying the (R, k) values ranging from (0.4, 1.8327) to (8, 1.0000) in steps of 0.2 atomic unit (Bohr) in R is also enclosed herewith. Using this list of (R, k) values, the values of Sab, Haa, Hab and U are obtainable as functions of the internuclear distance R (using the aforementioned expressions). Now from the U vs. R plot (as shown in figure), quite realistic values for the bond-length (equilibrium internuclear distance) Re (i.e., the value of R for which U is minimum) and for the equilibrium bond-dissociation energy De (the difference between U values at R = Re and at R = a) may be obtained.
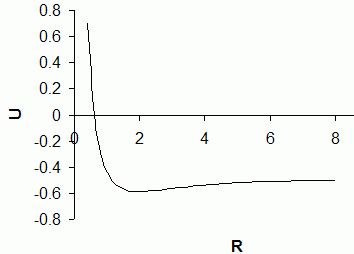
Procedure: This set of long and complicated calculation requires the use of a programmable calculator, such as the Assam-Calcu computer-software (available free of cost from many sources within the Internet). Within the Assam-Calcu command interface, one first runs the commands R = 0.4 and k = 1.8327, and then runs (after duly typing in) the above three expressions for Sab, Haa and Hab one by one, and finally the aforementioned expression for U, thereby getting the value (0.7012) of U for R = 0.4. Next, one would similarly enter R = 0.6 and the corresponding appropriate (listed) value for k into Assam-Calcu, and then would just re-run the last four commands (either as recallable individual old commands, or better via the appropriate re-run command which may be generated by invoking the corresponding wizard) regarding calculation of Sab, Haa, Hab and U. The new value of U (-0.0001) for R = 0.6 is thus obtained. Now one needs to repeat the above process (the same re-run command may be just recalled as an old command and used, there's no need to invoke the command-generating wizard again) for all other values of R (starting from 0.8 and up to 8.0) for which k is available within the aforesaid list. {To jot down current values of all the variables (R, k, Sab, Haa, Hab, U) from one screen, the List Variables button should be clicked.} After these calculations are over, the required plot of U vs. R is to be drawn, and then the Re and the De value estimated therefrom.