Medical Pharmacology Topics
![]()
![]()
| Preliminary Outline |
| Antimetabolite
Agents |
Antimetabolite agents are structuraly similar to physiological intermediates thus can act as substrates in biochemical reactions. They interphere with the production of nucleic acids by one or both of two major mechanisms: inhibition of dNTP synthesis or incorporation into DNA.
There are two major de novo pathways for the synthesis of nucleotides: purine synthesis and pyrimidine synthesis. An antimetabolite can inhibit one or more steps in either of these pathways.
Purine-Like Antimetabolites
The purine nucleotide synthesis pathway can be inhibited at 4 steps by antimetabolites:
 Addition
of the amine group precursor to the purine base by phosphoribosyl pyrophosphate
(PRPP) amidotransferase is inhibited by 6-mercaptopurine and 6-thioguanine
Addition
of the amine group precursor to the purine base by phosphoribosyl pyrophosphate
(PRPP) amidotransferase is inhibited by 6-mercaptopurine and 6-thioguanine6-mercaptopurine must be converted to 6-thioinosine-5-phosphate (T-IMP) to be active. T-IMP is a poor substrate for guanylyl kinase, the enzyme that converts GMP to GTP, thus accumulates in cells. Elevated T-IMP levels inhibit de novo purine synthesis by inhibiting PRPP amidotransferase.
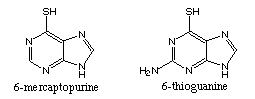
6-thioguanine works by a mchanism similar to 6-mercaptopurine. It is also converted to a ribonucleotide, which inhibits DNA synthesis. incorporation into DNA and RNA may also be an important determinant of cytotoxicity.
 6-mercaptopurine
is inactivated by xanthine oxidase, therefore the dose must be hal in patints
taking allopurinol. It is avaiable in both oral and intravenous forms. Allopurinol
is sometimes added to 6-mercaptopurine regimes to prevent hyperuricemia (?).
6-mercaptopurine
is inactivated by xanthine oxidase, therefore the dose must be hal in patints
taking allopurinol. It is avaiable in both oral and intravenous forms. Allopurinol
is sometimes added to 6-mercaptopurine regimes to prevent hyperuricemia (?).
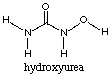
Hydroxyurea inhibits ribonucleotide reductase and thus blocks the conversion of ribonucleotide disphosphates to deoxyribonucleotides. It is eliminated by the kidneys and may cause skin rashes and occasional renal and hepatic toxicity. Hydroxyurea is used to treat chronic granulocytic leukemia. Since it decreases the pool of dNTPs, it may useful to combine this drug with inhbitors of DNA repair and DNA damaging agents.
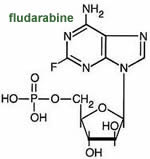 Fludarabine
is a purine antimetabolite that is phosphorylated inside the cell to the active
agent 2-fluoro-ara-ATP. This metabolite appears to inhibit DNA polymerase a,
ribonucleotide reductase and DNA primase, thus inhibiting DNA synthesis. Fludarabine
is useful fr treating chronic lymphocytic leukemia and low grade lymphomas.
It is available a IV injection or capsules.
Fludarabine
is a purine antimetabolite that is phosphorylated inside the cell to the active
agent 2-fluoro-ara-ATP. This metabolite appears to inhibit DNA polymerase a,
ribonucleotide reductase and DNA primase, thus inhibiting DNA synthesis. Fludarabine
is useful fr treating chronic lymphocytic leukemia and low grade lymphomas.
It is available a IV injection or capsules.
The renal clearance of fludarabine is about 40% of the total clearance. Patients with moderate impirment of renal function should have a 20 % dose reduction. It should not be administered to patients with severe renal impairment. Fludarabine should not be used in combination with pentostatin due to the risk of severe pulmonary toxicity.
The degree and severity of fludarabine's side effects depend on the amount and schedule of adminisration. High doses have been associated with irreversible CNS toxicity characterized by delayed blindness, coma and death.
Deoxycoformycin inhibits the purine nucleotide degradation pathway enzyme adenine deaminase, and leads to increased levels of dATP. It is not clear how this produces cytotoxicity. Deoxycoformcin is eliminated renaly and may produce neurological and renal toxicities.
 Pyrimidine-Like
Antimetabolites
Pyrimidine-Like
Antimetabolites
Pyrimidine nucleotide synthesis may be inhibited at three sites:
5-Fluorouracil was designed ant synthetized by Heidelberger after noticing that tumor cells require large amounts of uracil. 5-fluorouracil is phosphorylated to thee triphosphate and incorporated into RNA. Production of deoxyFUMP inhibits thymidilate synthase, trapping the enzyme in an intermediae step.
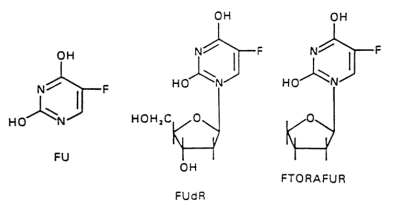
5-fluorouracil is extensively metabolized by the liver, thus must be administered IV. Long continuous infusion seem to work better than rapid injection. When used to treat liver cancer, the hepatic artery can be cannulated and 5-fluorouracil administered directly into the liver via a subcutaaneous infusion pump. Measurement at the hepatic vein demonstrate > 90% extraction by the liver. Weekly administration is not associated with significant toxicity other than the usual GI symptoms and myeolosupression.
5-fluorouracil is also used to treat cancers of the colon, stomach, breast, head and neck. Intraperitonal delivery is used to treat ovarian cancer. A topical preparation is available for basal cell skin cancer.
 Cytosine
arabinoside (Ara-C) differs from cytidine only for the orientation of one hydroxyde.
It may be converted to the pharmacologically inactive Ara-U by cytidine deaminase
or Ara-CMP by deoxycytidine kinase. Ara-CMP can then be deactivated to Ara-UMP
by dCMP deaminase or to Ara-dCTP by consecutive catalysis of dCMP kinase and
NDP kinase. The retention of this drug is an important determinat of cytotoxycity,
and will depend on the balance between deaminases and kinases.
Cytosine
arabinoside (Ara-C) differs from cytidine only for the orientation of one hydroxyde.
It may be converted to the pharmacologically inactive Ara-U by cytidine deaminase
or Ara-CMP by deoxycytidine kinase. Ara-CMP can then be deactivated to Ara-UMP
by dCMP deaminase or to Ara-dCTP by consecutive catalysis of dCMP kinase and
NDP kinase. The retention of this drug is an important determinat of cytotoxycity,
and will depend on the balance between deaminases and kinases.

The triphosphate inhibits DNA polymerase during replication and repair of UV-induced damage. In addition, it can be incorporated into DNA and inhibit template function and RNA elongation.
The half-life of cytosine arabinoside is 7-20 min, thus continuous infusion or large bolus will be more effective than smaller doses. Cytotoxicity is related top duration of exposure. A complete remision of leukemia is more probable when the drug is given by continuous infusion. The more Ara-CTP is incorporated into DNA, the less likely the acute myelogenus leukemia blasts will survive. Ara-CTP can only be incorporated during S phase, hus cells not in S phase will be resistant.
At high doses, Ara-C may cause brain and liver toxicity. In induction therapy for acute non-lymphocytic leukemia, Ara-C is usually given in combination with daunomycin or adriamycin. The usual treatment is continuous infusion for 7 days, or intrathecal injection to treat carcinomatous and lymphomatous meningitis.
 Gemcitabine
is a pyrimidine antimetabolite similar to Ara-C. It is converted to the active
forms inside the cell: gemcitabine diphosphate and gemcitabine triphosphate.
The diphosphate inhibits ribonucelotide reductase (converts nucleotides into
deoxynucleotides), inducing a depletion of the cellular pool of deoxynucleotides.
Depletion of dCTP also stimulates phosphorylation of the diphosphate and incorporation
of gemcitabine nucleotides into DNA. Gemcitabine triphosphates mediate inhibition
of dCMP deaminase (converts dCMP to dUMP), and this is responsible for the long
half-life and good activity of gemcitabine in solid tumors.
Gemcitabine
is a pyrimidine antimetabolite similar to Ara-C. It is converted to the active
forms inside the cell: gemcitabine diphosphate and gemcitabine triphosphate.
The diphosphate inhibits ribonucelotide reductase (converts nucleotides into
deoxynucleotides), inducing a depletion of the cellular pool of deoxynucleotides.
Depletion of dCTP also stimulates phosphorylation of the diphosphate and incorporation
of gemcitabine nucleotides into DNA. Gemcitabine triphosphates mediate inhibition
of dCMP deaminase (converts dCMP to dUMP), and this is responsible for the long
half-life and good activity of gemcitabine in solid tumors.
The distribution of gemcitabine's ative metabolites is limited by saturable process, thus giving an excess of drug will only be wasteful, as it will be eliminated before convertion to the active metabolites. Gemcitabine is deactivated in the liver by deamination to an inactive uracil metabolite. The most important side effect is low white blood cell counts. It is useful for treating pancreatic, lung, breast, ovarian and bladder cancers.
Methotrexate
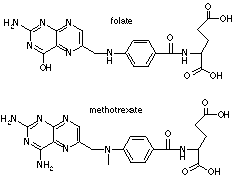 Methotrexate
resembles folic acid, therefore it is taken up by the folate carrier and binds
reversibly to DHFR. Both methotrexate and folate are polyglutamated by polyglutamate
reductase, confering water solubility and thus requiring the folate transporter
to enter the cell. Trimetrexate is not polyglutamated and remains lipid soluble,
thus can diffuse inside the cell without the need for a transporter. Trimetrexate
can be useful to treat transport-deficient methotrexate-resistant tumors.
Methotrexate
resembles folic acid, therefore it is taken up by the folate carrier and binds
reversibly to DHFR. Both methotrexate and folate are polyglutamated by polyglutamate
reductase, confering water solubility and thus requiring the folate transporter
to enter the cell. Trimetrexate is not polyglutamated and remains lipid soluble,
thus can diffuse inside the cell without the need for a transporter. Trimetrexate
can be useful to treat transport-deficient methotrexate-resistant tumors.
However, polyglutamated methotrexate has a higher affinity for thymidilate synthase (converts dUMP to dTMP) and cannot leave the cell. Accumulation of polyglutamated methotrexate inside the cell leads to accumulation of dihydrofolate and subsequent conversion to formyldihydrofolates, which depleates the pool of usable folate. Thus the cytotoxicity of methotrexate is two-fold in the purine synthesis pathway: DHFR inhibition and folate depletion.
Excess tetrahydrofolate can rescue both neoplastic and normal cells from the cytotoxicity of methotrexate. Other mechanisms of resistance to methotrexate include a deficient folate transporter and DHFR mutations that reduces its affinity for methotrexate.
Methotrexate binds t lasma proteins, thus other drugs that bind to plasma proteins may potentiate its toxicity: aspirin, sulfonamides, penicillins, etc. I is distributed to total body water and filtered, secreted, and reabsorbed by the kidneys. It is excreted as a salt of a weak acid, thus other drugs also excreted this way, lik aspirin and penicillins, will interfer with methotrexate excretion. At high dose, it will penerate the CNS.
High dose treatment with methotrexate with leucovorin rescue is used for resistant tumors. Leucovorin competes wih methotrexate for the folate transporter. Success of this therapy requires neoplastic cells to lack the transporter (?). If normal cells have polyglutamated methotrexate, leucovorin will probabli not rescue them. Bone marrow cells do not form polyglutamates, therefore they are not affected by methotrexate.
At high IV doses, methotrexate can cause renal failure, neurotoxicity and hepatic failue. Intrathecal administration may cause neurotoxicity or leukoencephalopathy in children.
Methotrexate is used IV to treat leukemia, chroriocarcinoma in females, head and neck cancrs, brest cancer, sarcoma and lymphoma. Intrathecal administration is used to treat carcinomatous meningitis. Oral methorexate is used to treat psoriasis although may cause cirrhosis in 25% of patients. May also be used to treat rheumatoid arthritis.
![]()
![]() Continue
to "Natural Anti-Tumor Agents" or take
a quiz: [Q1].
Continue
to "Natural Anti-Tumor Agents" or take
a quiz: [Q1].
Need more practice? Answer the review questions below.
Questions coming soon